04 Visualizing and counting of the alignments
Overview
Teaching: 45 min
Exercises: 15 minQuestions
What is a BAM file?
What do all these numbers mean?
how to prepare BAM files for visualisation?
How to use IGV, an interactive genome browser?
Objectives
Learn how to upload and view your BAM alignment file in IGV.
Create a count file to extract the number of reads that maps within a gene.
Table of contents
1. The SAM/BAM format
SAM files, are tab-delimited text files that contain information for each individual read and its alignment to the genome. While we do not have time to go in detail of the features of the SAM format, the paper by Heng Li et al. provides a lot more detail on the specification.
The compressed binary version of SAM is called a BAM file. We use this version to reduce size and to allow for indexing, which enables efficient random access of the data contained within the file.
1.1 What’s in a SAM/BAM file
The file begins with a header, which is optional. The header is used to describe source of data, reference sequence, method of alignment, etc., this will change depending on the aligner being used. Following the header is the alignment section. Each line that follows corresponds to alignment information for a single read. Each alignment line has 11 mandatory fields for essential mapping information and a variable number of other fields for aligner specific information. An example entry from a SAM file is displayed below with the different fields highlighted.
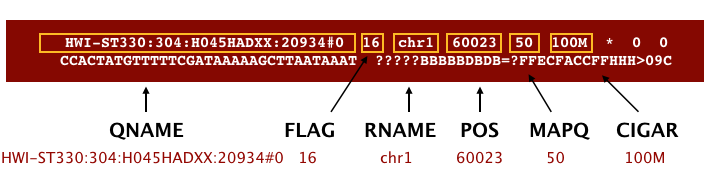
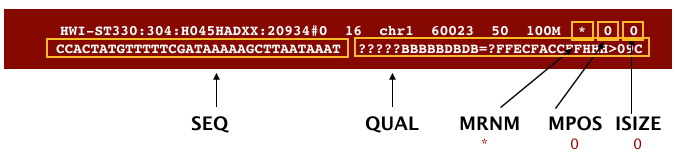
Additionally tags (or attribute) can be aded to each of of the lines. These tags give some aditional information on the alignment. The number and type of tags varies between different alinment tools and the settings within these tools.
Here a list of tags that are commonly used.
| Tag:Type | Meaning |
|---|---|
| NM:i | Edit distance |
| MD:i | Mismatching positions/bases |
| AS:i | Alignment score |
| BC:z | Barcode sequence |
| X0:i | Number of best hits |
| X1:i | Number of suboptimal hits found by BWA |
| XN:i | Number of ambiguous bases in the reference |
| XM:i | Number of mismatches in the alignment |
| XO:i | Number of gap opens |
| XG:i | Number of gap extentions |
| XT | Type: Unique/Repeat/N/Mate-sw |
| XA:z | Alternative hits; format: (chr,pos,CIGAR,NM;) |
| XS:i | Suboptimal alignment score |
| XF:i | Support from forward/reverse alignment |
| XE:i | Number of supporting seeds |
To start of we’ll have a look at how to use samtools to have a peak at the the contents of the bam files.
As these file are binary you can not simply use:
$ head Arabidopsis_sample1.bam
?BC?mRK??0t
=8?W???F?????BlӔ?^;?n
?#??ٟ۟T??CA??h?????????$?|?K??
?????чa??z?9|M???4??~WH??5??7???ǻ?Ԇr?~wE?Bd"???}q????.??c?K^?}?GM?s???@(}??ql&R??=RF?H??I???9?????Q:5eM?M?4?ጃ3??=??7?^?+x????sp
??
8??$???0?g?V?Xy?!???hm?#?P2?qX?`<t??? -?<???????
?@?81??
? ???+?
c:??G?! H
???v*???І?Pv???c?x????y?a)?/??S?8[??ޒ?y!?P:,??-5??????֫$I^ǽ???ͧ_?ߗ??<??BChc?\k?eYU????a???kw?}????}??????8?:???!?
3(
QD??(?T??p?C??
?D?"?ф 0?? F???? ?0h&d?o?}έ?==u?F?tUݺU???k?????o??F???q????v????U????A????p{????ޕg?^p?ht?n?zj4j?W{L?mٕ??
!M)J???n:?3M??*5???U?>P???!???ٍi?$I?eY?-
c???0
?H????? =?R?}WG/~>??????ޫ????s??/l?^r?/z??????x^u???'?'U?x@???N`o??G#?m??P?)ӕ?S??U?H?5?ѕ?\xJy???NH??????
?f?>?R?I
This will give an unreadable result. SAMtools can help us to make the content readable.
1.2 SAMtools
SAMtools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format.
Like many Unix commands, samtools commands follow a stream model, where data runs through each command as if carried on a conveyor belt. This allows combining multiple commands into a data processing pipeline. Although the final output can be very complex, only a limited number of simple commands are needed to produce it. If not specified, the standard streams (stdin, stdout, and stderr) are assumed. Data sent to stdout are printed to the screen by default but are easily redirected to another file using the normal Unix redirectors (> and >>), or to another command via a pipe (|).
1.3 SAMtools commands
SAMtools provides the following commands, each invoked as “samtools some_command”.
- view
The view command filters SAM or BAM formatted data. Using options and arguments it understands what data to select (possibly all of it) and passes only that data through. Input is usually a sam or bam file specified as an argument, but could be sam or bam data piped from any other command. Possible uses include extracting a subset of data into a new file, converting between BAM and SAM formats, and just looking at the raw file contents. The order of extracted reads is preserved. - sort
The sort command sorts a BAM file based on its position in the reference, as determined by its alignment. The element + coordinate in the reference that the first matched base in the read aligns to is used as the key to order it by. [TODO: verify]. The sorted output is dumped to a new file by default, although it can be directed to stdout (using the -o option). As sorting is memory intensive and BAM files can be large, this command supports a sectioning mode (with the -m options) to use at most a given amount of memory and generate multiple output file. These files can then be merged to produce a complete sorted BAM file [TODO - investigate the details of this more carefully]. - index
The index command creates a new index file that allows fast look-up of data in a (sorted) SAM or BAM. Like an index on a database, the generated *.sam.sai or *.bam.bai file allows programs that can read it to more efficiently work with the data in the associated files. - tview
The tview command starts an interactive ascii-based viewer that can be used to visualize how reads are aligned to specified small regions of the reference genome. Compared to a graphics based viewer like IGV,[3] it has few features. Within the view, it is possible to jumping to different positions along reference elements (using ‘g’) and display help information (‘?’). - mpileup
The mpileup command produces a pileup format (or BCF) file giving, for each genomic coordinate, the overlapping read bases and indels at that position in the input BAM files(s). This can be used for SNP calling for example. - flagstat
Counts the number of alignments for each FLAG type.
Looking at the content of the file using samtools view:
$ samtools view Arabidopsis_sample1.bam | head
ERR1406259.2 16 Chr1 17425094 60 85M228N16M * 0 0 TGAGAATAAAACCATATGGGTTGGTGATTTGCATCACTGGATGGATGAGGCTTATCTTAATTCTTCTTTTGCTTCCGGCGACGAGAGAGAGATTGTTTCGG CDEDDDDDB@4DEDDDDDDDDDDDDDDDDDDDDDCAEEDEFFFEFFHHHGHJJJIHJJJIIIGIIJIJJJJJJJFJJJIIIGJJIJJJHHHHHFFFFFCCC AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU XS:A:+ NH:i:1
ERR1406259.5 0 Chr1 25446585 60 101M * 0 0 AATTATTGGGCCATATCCCGACCCCTTTGGCAAACCCATCGACCGTTCCAAGAATCCCTCCGGTAATCCCTCCGGCAACCCCAATAATAAGCTTATCAAGC CCCFFFFFHHHHGJIJJJJJJJJIJJJIIIDHJIIJJJJJGIJGIFGIGIHHHHHHFFFFDDDB?BD@CDDDBBDDDDDDDDDDDDDEEDDDDDDDDEDDC AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU NH:i:1
ERR1406259.11 0 Chr1 22700646 60 101M * 0 0 GCCATGTTGGGTGCAGCTGGAGCTATTGCTCCTGAGATTTTAGGAAAGGCTGGTCTGATTCCAGCAGAGACTGCTCTTCCTTGGTTCCAAACCGGTGTGAT @@@DFDDEFDHBFHIIJJGIIJJIIJJFJFJJJJJJGIJJJJIGGHJ@FGHIE=FHIJJIGEHGJJGH=A3=CED?DEFF<CCCCCDCEDDDDBD385@DD AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU NH:i:1
ERR1406259.10 0 Chr1 11215252 60 69M263N32M * 0 0 CCGTTCACTATCAAGTGCGGTAAACCGTCTGACCCGCATAACAATCCGTACTTCCCGTAGTTGTCGAACCTGCGTTTGGTTTTCTCGATCTGAGCATTGAG @@CFFFFFHHHHHJJBGIHIFFIJJGGIIIIJIGGGIIIIJIIGIIJIGIIGGJJHHHFFBDEECBA;ABCDDD289@D<?BB>@AB:>BBD>>>CCC@>C AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU XS:A:- NH:i:1
ERR1406259.12 16 Chr1 30360823 60 101M * 0 0 AAGAGTGTCACAGAGCTTGAGAAGGAATATGAGATTAGGGGCTGCGGCAGAAAGGACTGGATTGATAAAAGAGGAGACTGGAGGTCTAAGGCTTATGGTTG CDDDDDCDCCCDCCDDEEEEEEFFDDFEFFHEEC;ACGHFGGFJJIIHGF?*JJIHGGJJIJIGHBJJIJIJJIHFBIHJJJIGGGJJHHHHHFDFFF@@? AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU NH:i:1
ERR1406259.13 0 Chr1 25048653 60 49M136N52M * 0 0 CATCATAGTATCCGGGTGAGTTACCGTGCTCACGGTACACAAATCCGTGCTCCAACTCGAATTCAACACAAGGAATCCACTTGTTGCGGATAAGGTAGTCA CCCFFFFFFHHHHJJJEFHJJIJJJIGIJJJJJJJGIIJJJJJJJJJJJJJJJJGIJJHHHHFFFFFFDDDEDDDCDDDDDDDDDDDDDDDDDDD@@CDED AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU XS:A:- NH:i:1
ERR1406259.17 16 Chr1 19517907 60 101M * 0 0 TCCAAGCTGAGGGAAGAACTCTAGATGATCAAGAATCCTATCGGGACAAAATAATTGAGTACAAGAGTCTTGAAGATGTTGTTGAGGATAATATCAGTTTG ADDDDDDDDEEEEEEFFDFFFHHHHHHHHJJJIIGGIJJJJJJJJJJJJJIJJJJJJJJJIJJIJIHIJJJJJJJJJJJJJJJJJJJJHHHHHFFFFFCCC AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU NH:i:1
ERR1406259.24 16 Chr1 6804452 60 49M153N52M * 0 0 CAGCATCGTCCACTTCCACTTGCCCTCCCGGCGGCAATAATTTGCACAACTGTGGGGCTACAAGAATGTAACCATGCGAAGCGATGTGGTTAAGAACGTCA :4?8(0>@CCC@CC@>>4<82<@<8=?8896=7E>EC?CD@IGGC=@C7CAF@@BDB9?9FHGD>BBHDFBCIIHHBGHEHBGA<+ACBBHDFDBA;D@<@ AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:101 YT:Z:UU XS:A:- NH:i:1
ERR1406259.25 0 Chr1 7738049 60 101M * 0 0 GCTGAACGAATGGCCCGGAGAAGCGATTTTGATCAGNATTCATCAGGAGGAGCCAGTTATCCATCACACGGTGAGATNTACGAAGTTGTTCTCTACTTTGG CCCFFFFFGFHHHJJJJJGGIJBBHIGIIIEAEDHI#0<FHEIEGIEGIICFCACE>BEFCCEDEEDCD@B?B@BCC#,8??@AB?CDDDDECCACDDEAC AS:i:-2 XN:i:0 XM:i:2 XO:i:0 XG:i:0 NM:i:2 MD:Z:36T40G23 YT:Z:UU NH:i:1
ERR1406259.32 16 Chr1 4393371 60 99M2S * 0 0 TAAGAACCTTCTTGGCTCCAGCCTGAAGGTGCTTCCCAGCACCGTCTCTGTCAACAAACACTCCGGTTCCTTCGATAACTAAGTCAATGCCTAGTTCCCCC ADCA>3BC?BDCDDDDBDDBDDDDCCCDDDDBCBDEEED?5GGHHIIJJJIGHIJHF=FCJHJJJJJIIIJJIJIJJJJIJJJIIIGJGHFFHFFFFFCC@ AS:i:-2 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:99 YT:Z:UU NH:i:1
SAMtools will make the data readeble, this data is then piped through head to show the first 10 lines of the file.
2.4 Counting and sorting
SAMtools view can be used to filter the alignment based on characters like mapping quality, chromosome, orientation etc. When the -c option is added the filtered selection is counted.
Count the total number of records:
$ samtools view -c Arabidopsis_sample1.bam
$ 263657
Count with flagstat for additional information:
$ samtools flagstat arabidopsis1.bam
263657 + 0 in total (QC-passed reads + QC-failed reads)
14232 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
263142 + 0 mapped (99.80% : N/A)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)
0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
Count the records using the FLAG argument. Count the alignments that don’t align.
$ samtools view -f 4 -c Arabidopsis_sample1.bam
$ 515
The argument -f includes reads that fit samflag 4, read unmapped.
Count the reads that do align:
$ samtools view -F 4 -c Arabidopsis_sample1.bam
$ 263657
Here -F is used to exclude reads that fit samflag 4, read unmapped. Everything else is included.
Question
Sometimes you will see that this number of alignments is higher then the number of sequences in your fastq file. How can this be?
Solution
When a read multimaps (aligned to multiple positions in the genome), each of these positions is included as a separate alignment.
Write your selection to a new file:
samtools view -Sb -F 4 -o Arabidopsis_sample1_mapped.bam Arabidopsis_sample1.bam
In this command -Sb is needed to keep the file binairy(compressed), and -o specifies the output filename (a bam file again).
Count the reads that align to the forward strand:
$ samtools view -F 20 -c Arabidopsis_sample1.bam
$ 131631
Use -F 20 to exclude “read reverse strand” and “read unmapped”.
Count the reads that align to the reverse strand:
$ samtools view -f 16 -c Arabidopsis_sample1.bam
$ 131511
With -f 16 you select for “read reverse strand”.
With SAMtools it is also posible to select for alignments with a minimal mapping quality.
Alignments with a maximal score (60 for hisat2 output files and 255 for STAR output files) are truly unique:
$ samtools view -q 60 -c Arabidopsis_sample1.bam
$ 240224
This number can never be bigger then the number of reads in the fastq file, as all reads in the output give a single alignment.
Question
BAM files usually contain a tag or attribute that gives the number of mismatches between the read and the reference genome. With SAMtools it is unfortunately not possible to filter on these values. Could you think of an other way to select for alignments that align without any mismatches?
Hint: make use ofgrep "XM:i:0"among others.Solution
samtools view Arabidopsis_sample1.bam | grep "XM:i:0" | wc -l
3. Visualization of a BAM file
A BAM file can be visualized using a genome viewer like IGV.
We can’t just upload the files in the viewer. We first need the files to be sorted and indexed.
This can be done making use of SAMtools sort and index commands.
3.1. Preparation of the BAM file for IGV
Sorting
Samtools can also be used to sort the read alignments. The aliments will be sorted based on the position ofv the alignment on the reference genome, starting from the beginning of chromosome 1 to the end of the last chromosome.
$ samtools sort -o arabidopsis1_sorted.bam arabidopsis1.bam
where -o defines the name of the output file (also a BAM file).
The default for samtools sort is sorting by position. There are more sorting posibilities to be found with samtools sort --help.
Indexing
An index file is needed to get access rapidly to different alignment regions in the BAM alignment file.
samtools index arabidopsis1_sorted.bam
Only the input file name needs to be specified, based on this name a .bai (BAM index) is produced.
Transfer the files to your local computer
Next the files need to be downloaded to your local computer.
First, we need to exit the container (ctrl/cmd + d) and next create a folder on your VM outside the container:
$ mkdir IGVfiles
Copy the files (both the .bam and the .bai), or just the whole content of the directory mapped:
$ docker cp bioinfo:/home/mapped/ ~/IGVfiles/
Go to your local computer, create a directory and download the files from the VM:
$ mkdir IGV
Download the sorted .sorted.bam files:
$ scp -r root@178.128.240.207:~/home/tutorial/IGVfiles/mapped/*sorted.bam ~/Desktop/IGV
And the index (.bai) files:
scp -r root@178.128.240.207:~/home/tutorial/IGVfiles/mapped/*.bai ~/Desktop/IGV
3.2. IGV
For this exercise we’ll be making use of an online version of IGV.
The Arabidopsis genome that we used for the mapping is available in this web app.
It can be found under “Genome -> A. thaliana (TAIR 10)”.
The BAM files can be added as a track. Choose “Tracks -> local file”. Select both the .bam file and the accompanying .bai.
You should get something like this:
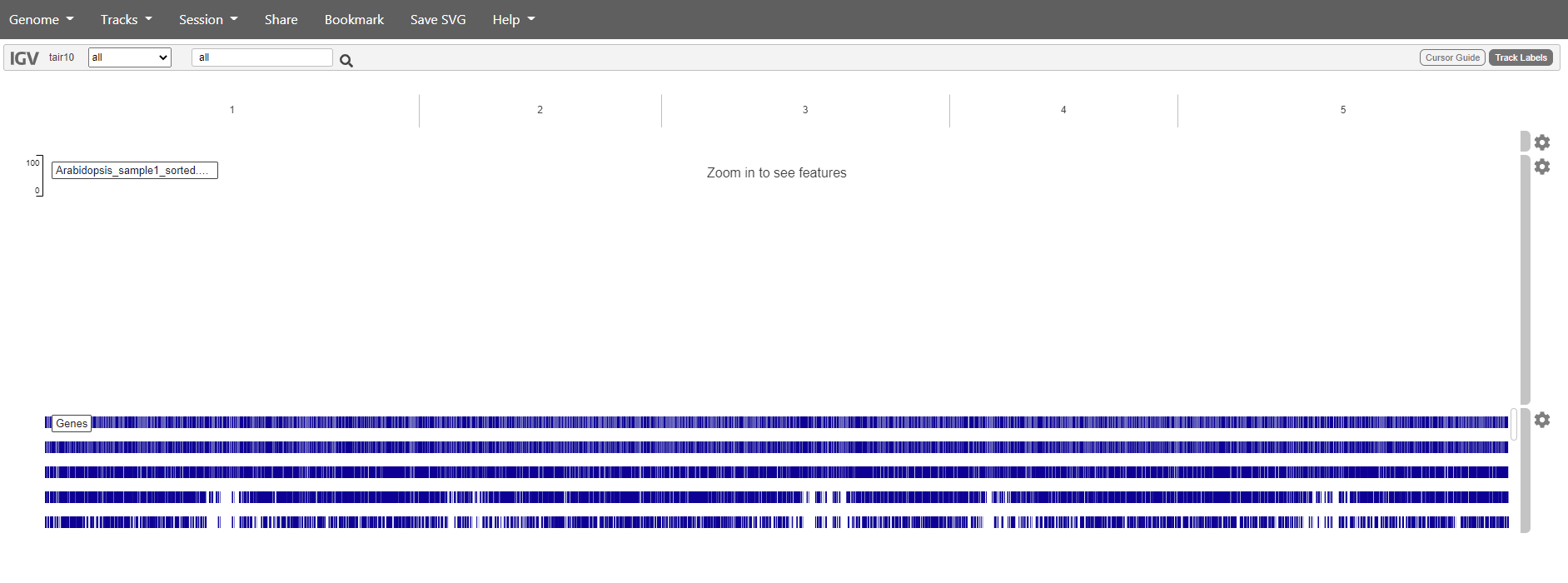
Mapping has in this case only been done against chromosome 1. So if we want to see are reads choose chromosome 1. Zoom in to see the reads.
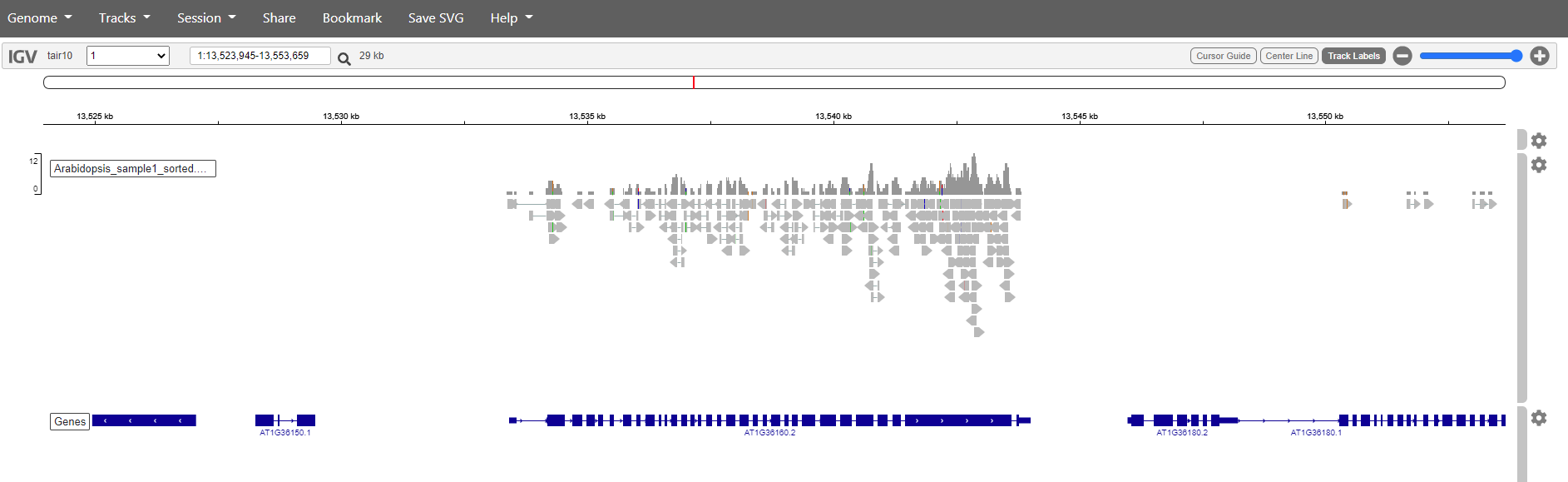
It is also posible to search for genes. Just write the name in the search box and click the magnification glass. Try with the genes:
- AT1G16080,
- AT1G10370
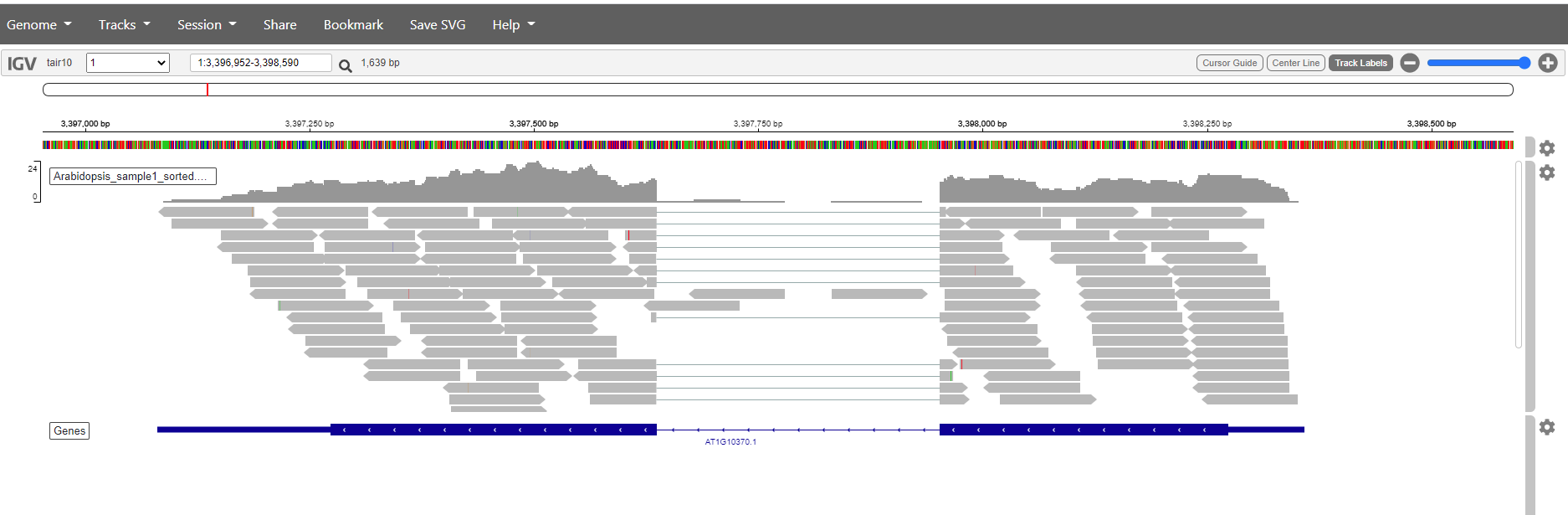
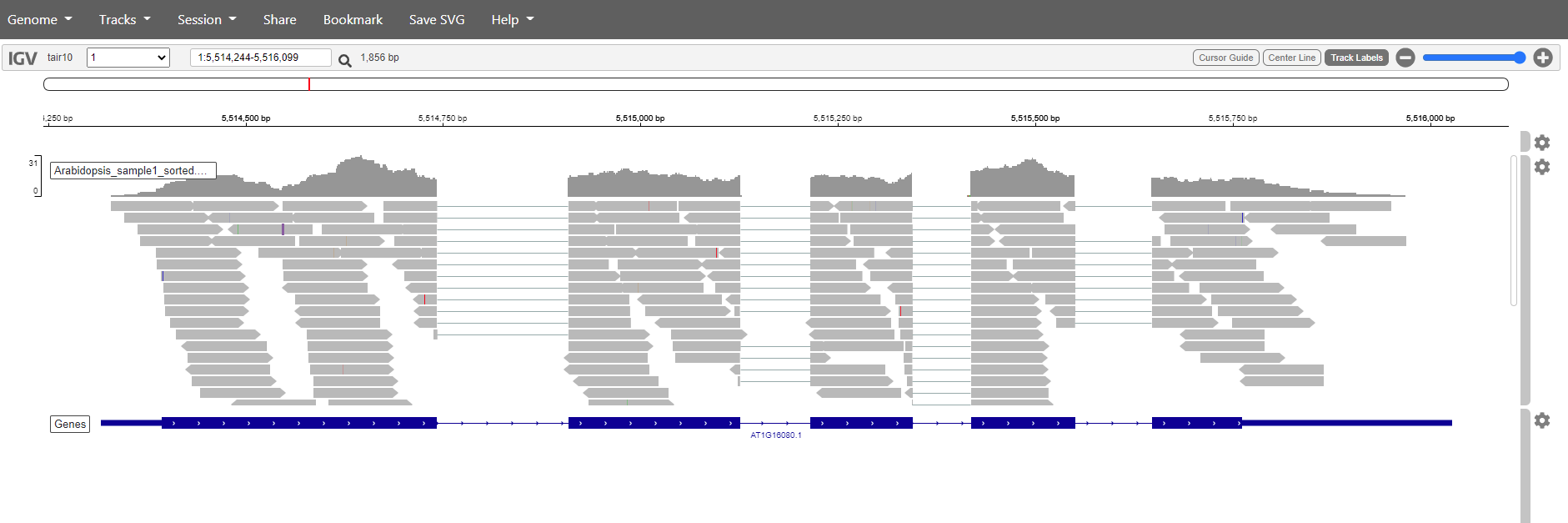
Question
Pick a sample and visualize the forward and reverse alignments separately in IGV.
Solution
Select and sort forward reads in one go.
samtools view -Sb -F 20 Arabidopsis_sample1.bam | samtools sort -o Arabidopsis_FW_sorted.bam
Index the forward reads.samtools index Arabidopsis_FW_sorted.bam
Select and sort the reverse reads.samtools view -Sb -f 16 Arabidopsis_sample1.bam | samtools sort -o Arabidopsis_RV_sorted.bam
Index the reverse reads.samtools index Arabidopsis_RV_sorted.bam
From the Desktop of your local computer create directory and download the files.mkdir IGVscp -r root@178.128.240.207:~/home/tutorial/IGVfiles/mapped/*sorted.bam* ~/Desktop/IGV
Upload the files in IGV and you should get something like this.
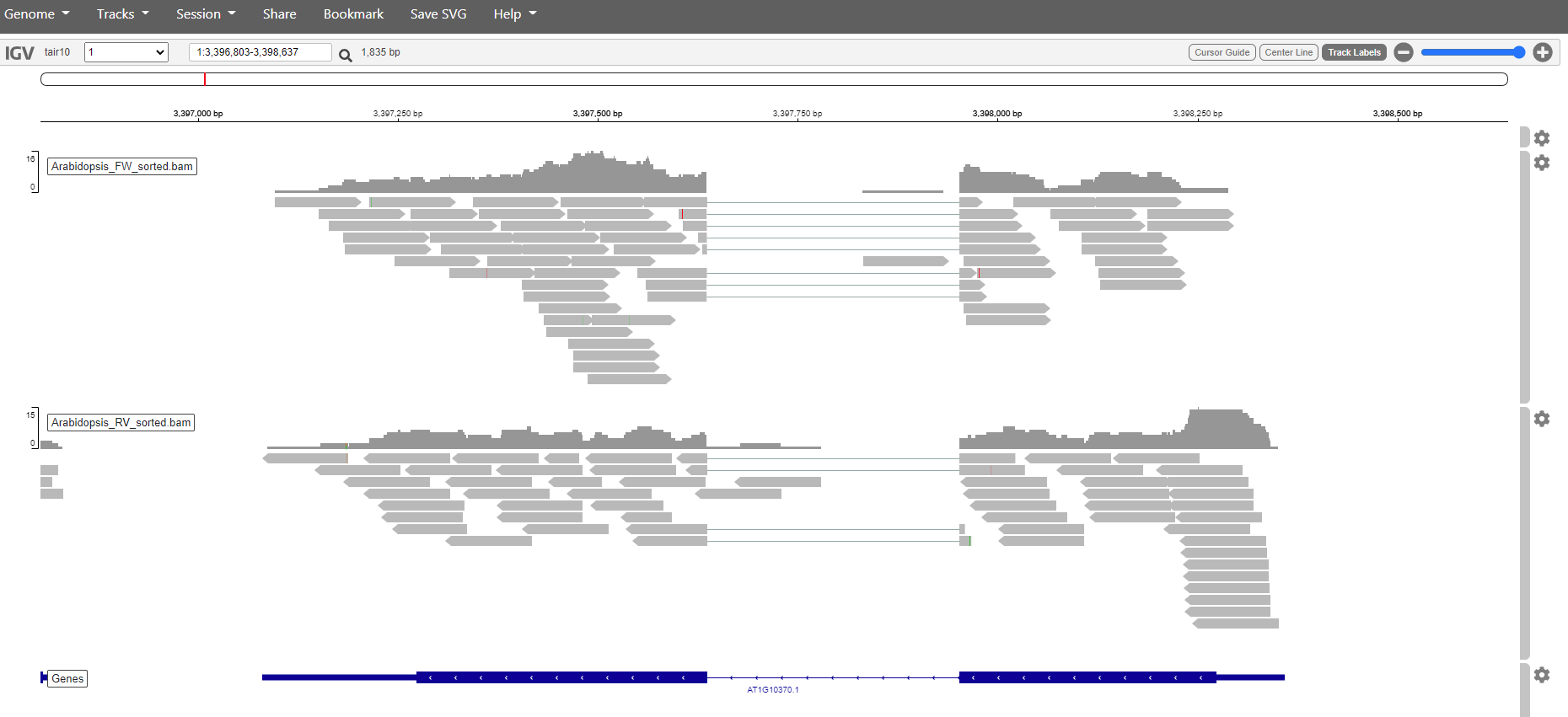
4. Creating the counts file
For downstream applications e.g. differential expression analysis, the number of reads that maps within a gene has to be determined for each sample.
The featureCounts program from the Subread package can do this. The complete user guide is available here and featureCounts is in section 6.2.
featureCounts can…count (!) the number of reads that map within a feature. The Arabidopsis genome annotation in the GFF3 format contain three different features to choose from.
Depending on the downstream applications the choice is gene, transcript or exon. In this study we are just looking for differentially expressed genes so our feature of interest specified by the -t will be gene.
$ cd /home/
$ gunzip ath_annotation.gff3.gz
$ featureCounts -O -t gene -g ID -a ath_annotation.gff3 -o counts.txt mapped/*.bam
Here is an explanation of the different arguments used:
-a <string>: Name of an annotation file. GTF/GFF format by default.-o <string>: Name of the output file including read counts.-O: Assign reads to all their overlapping meta-features.-t <string>: Specify feature type in the GTF/GFF annotation to summarise the counts.-g <string>: Specify attribute type in GTF/GFF annotation. This GTF/GFF determines the name of the features.
The output file produced by featureCounts is a tab-delimited file that can be opened in a spreadsheet program like Excel.
Key Points
The SAM/BAM format is the end-result of a read alignment to a reference genome.
The samtools software can be used to view, filter and order aligments in a .bam file.
The aligments can be visualized in a genome using an genome viewer like IGV
The resulting BAM files are used to generate a count table for use in differential expression analyses.