cd <path_to_software_folder>
git clone https://github.com/deweylab/RSEM.git
mv RSEM/ RSEM_1.3.3
cd RSEM_1.3.3
make
make install prefix=/<path_to_software_folder>/RSEM_1.3.3RSEM
Introduction
RSEM is a software package for estimating gene and isoform expression levels from RNA-Seq data (Li and Dewey 2011). The RSEM package supports threads for parallel computation of the EM algorithm, single-end and paired-end read data, quality scores, variable-length reads and RSPD estimation. In addition, it provides posterior mean and 95% credibility interval estimates for expression levels. For visualization, it can generate BAM and Wiggle files in both transcript-coordinate and genomic-coordinate. Genomic-coordinate files can be visualized by both UCSC Genome browser and Broad Institute’s Integrative Genomics Viewer (IGV). Transcript-coordinate files can be visualized by IGV. RSEM also has its own scripts to generate transcript read depth plots in pdf format.
For a full list of options, we recommend that the users visits the documentation and the tutorial.
Installation
Installed on crunchomics: Yes,
- RSEM v1.3.3 is installed as part of the bioinformatics share. If you have access to crunchomics and have not yet access to the bioinformatics you can send an email with your Uva netID to Nina Dombrowski.
- Individual RSEM modules can be found in this folder
/zfs/omics/projects/bioinformatics/software/RSEM_1.3.3/bin/
If you want to install it yourself, you can run:
Usage
There are many different options how to use RSEM as indicted in the figure below:
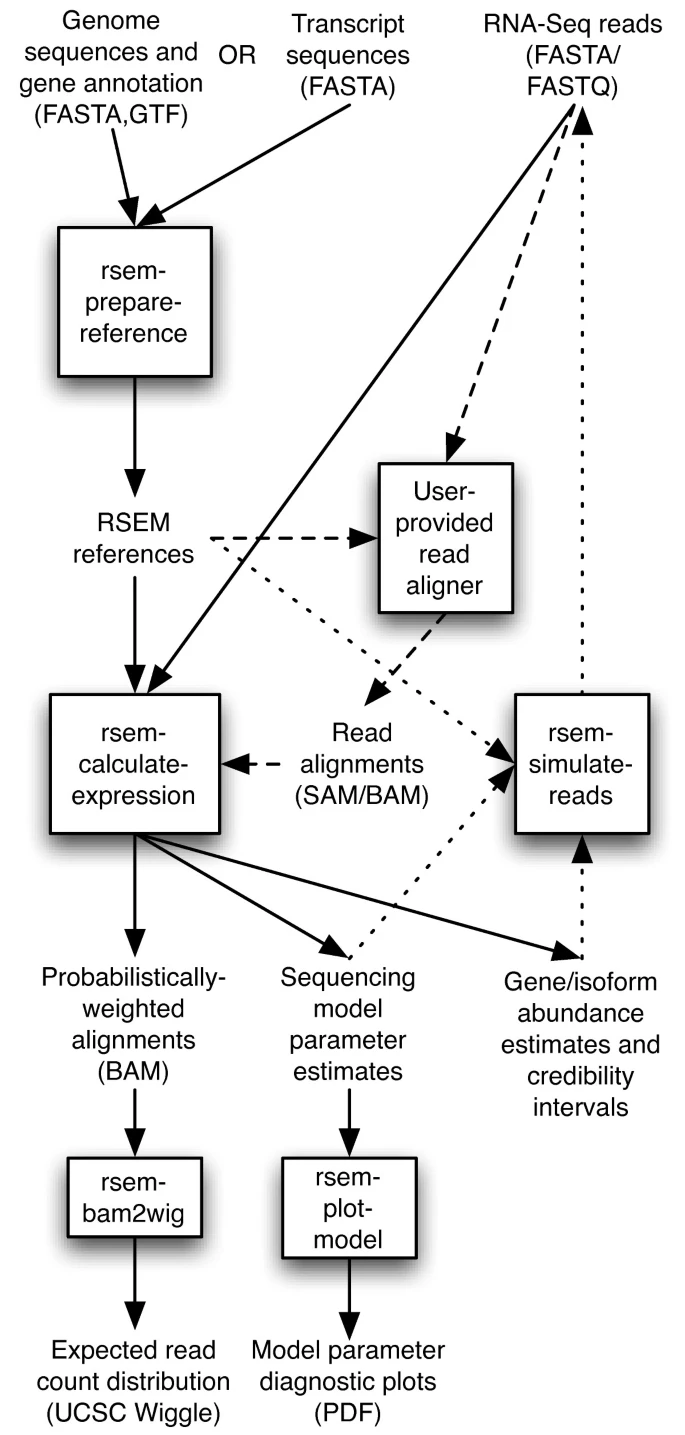
For a full list of options, we recommend that the users visits the documentation and the tutorial.
Below, you find a very brief example on how to use RSEM to estimate gene and isoform expression with BAM mapping files that were generated with STAR using --quantMode TranscriptomeSAM. Notice, you don’t need to run the mapping with an aligner separately, as RSEM supports read mapping with Bowtie2, STAR and HISAT2.
#prepare folders
mkdir -p data/genome_files/rsem_ref
mkdir -p results/quantification/rsem
#prepare the reference files
#for this step we need the fasta and gtf files from a reference assembly (the same assembly that was also used when running STAR)
/zfs/omics/projects/bioinformatics/software/RSEM_1.3.3/bin/rsem-prepare-reference \
--gtf data/genome_files/assembly.gtf \
data/genome_files/assembly.fasta \
data/genome_files/rsem_ref
#caculate the expression
#for this we need the bam files generated using STAR
/zfs/omics/projects/bioinformatics/software/RSEM_1.3.3/bin/rsem-calculate-expression \
--bam --no-bam-output -p 12 --paired-end --forward-prob 0.5 \
results/star_output/sample1_Aligned.toTranscriptome.out.bam \
data/genome_files/rsem_ref \
results/quantification/rsem/sample1 >& results/quantification/rsem/sample1.log
#if you ran star and rsem on more than one sample via a for-loop, then you can use the code below to
#combine rsem results for multiple samples
python3 /zfs/omics/projects/bioinformatics/scripts/combine_rsem.py \
-i 06_mapping/quantification/rsem \
-o 06_mapping/quantification/rsem/rsem_genes.tsv \
-t genes
python3 /zfs/omics/projects/bioinformatics/scripts/combine_rsem.py \
-i 06_mapping/quantification/rsem \
-o 06_mapping/quantification/rsem/rsem_transcripts.tsv \
-t isoformsOptions used in the example (please read the manual for a full set of options!):
--paired-endis applicable to paired stranded RNA-seq data--forward-probNUM. Here:- 0 is for a strand-specific protocol where all (upstream) read are derived from the reverse strand
- 1 is for a strand-specific protocol where all (upstream) reads are derived from the forward strand
- 0.5 is for a non-strand specific protocol
A note on combining results from different samples
We provide a small python script o combine the rsem outputs from multiple files. This script can be found on Crunchomics but you are free to combine the files yourself in your favorite computational language. The script combine_rsem.py requires that the gene and isoform files are located in a specific folder. Namely, each sample should be have a folder that has the same name as the sample. In the example above, the script expects a folder called sample1 in the target folder specified in the script, i.e. results/quantification/rsem/. In the sample1 folder, the script will look for two files, rsem.genes.results and rsem.isoforms.results.
If you want to use DESeq2, then you can import the individual tables into R with tximport.
References
Li, Bo, and Colin N. Dewey. 2011. “RSEM: Accurate Transcript Quantification from RNA-Seq Data with or Without a Reference Genome.” BMC Bioinformatics 12 (1): 323. https://doi.org/10.1186/1471-2105-12-323.