mamba create -n samtools_1.19.2
mamba install -n samtools_1.19.2 -c bioconda samtools=1.19.2
mamba activate samtools_1.19.2Samtools
Introduction
SAMtools is a toolkit for manipulating alignments in SAM/BAM format, including sorting, merging, indexing and generating alignments in a per-position format (Danecek et al. 2021). The SAM format is a standard format for storing large nucleotide sequence alignments and is generated by many sequence alignment tools such as Bowtie or BWA. The BAM format is the binary form from SAM.
For more detailed information, please visit the manual.
Installation
Installed on crunchomics: Yes, samtools v1.9 is installed.
If you want to install it yourself, the command below installs the version verified on Crunchomics. Before running it, check the htslib downloads page for a newer version and swap the version number if one is available. We install via bioconda rather than building from source, since bioconda keeps pace with the latest releases and this keeps the install simple.
Usage
The basic usage of SAMtools is:
samtools COMMAND [options]
The following commands are available:
- view: SAM/BAM and BAM/SAM conversion
- sort: sort alignment file
- mpileup: multi-way pileup
- depth: compute the depth
- faidx: index/extract FASTA
- tview: text alignment viewer
- index: index the alignment
- idxstats: generate BAM index stats
- fixmate: fix mate information
- flagstat: simple stats
- calmd: recalculate MD/NM tags and
=bases - merge: merge sorted alignments
- rmdup: remove PCR duplicates
- reheader: replace BAM header
- cat: concatenate BAMs
- bedcov: read depth per BED region
- targetcut: cut fosmid regions
- phase: phase heterozygotes
- bamshuf: shuffle and group alignments by name
For detailed description and more information on a specific command, just type:
samtools COMMAND
Below, you find examples on how to run some of the most common samtools commands. For this, we start with the example of a researcher, who aligned Illumina reads from a sample to a reference genomes and generated a SAM file using Bowtie 2.
What are SAM files
The SAM file is a tab-delimited text file that contains information for each individual read and its alignment to the reference.
The file begins with an optional header. The header describes the source of data, reference sequence, method of alignment,and might look slightly different depending on the aligner being used. Each section begins with character @ followed by a two-letter record type code. These are followed by two-letter tags and values.
Afterwards, you see the alignment section. Each line corresponds to the alignment information for a single read. Each alignment line has 11 mandatory fields for essential mapping information and a variable number of other fields for aligner-specific information. This looks something like this:
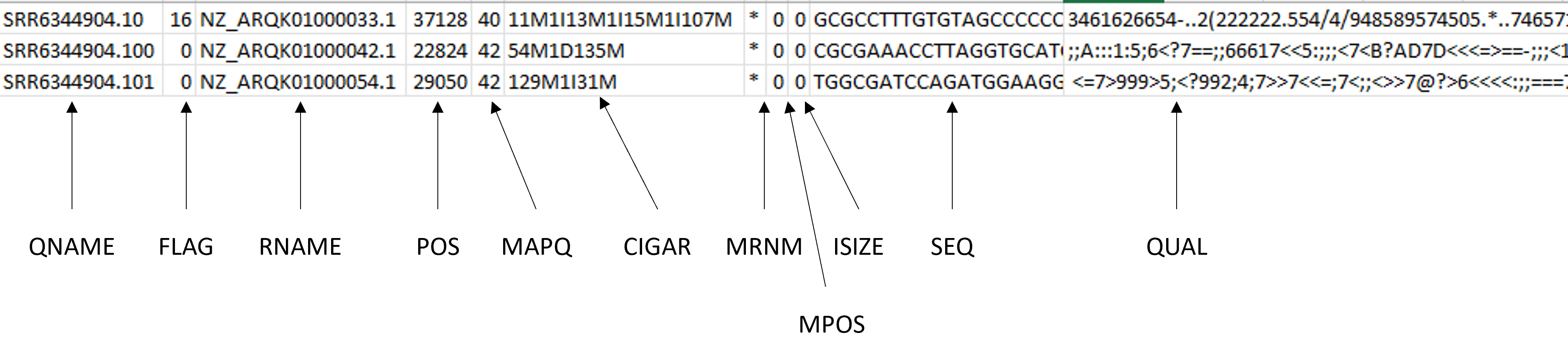
The individual fields are:
- QNAME: Query name or read name, the same read name present in the header of the FASTQ file
- FLAG: numerical value providing information about read mapping and whether the read is part of a pair. To translate these flags into a more meaningful description, go here. In our example:
- The 16 flag means that the short sequence maps on the reverse strand of the reference genome.
- The 0 flag means that none of the bit-wise flags you see in the link are set. This means that your reads with flag 0 are unpaired (because the first flag, 0x1, is not set), successfully mapped to the reference (because 0x4 is not set) and mapped to the forward strand (because 0x10 is not set)
- RNAME: the reference sequence name
- POS: refers to the 1-based leftmost position of the alignment
- MAPQ: the alignment quality, the scale of which will depend on the aligner being used. The maximum MAPQ value that Bowtie 2 generates is 42. In contrast, the maximum MAPQ value that BWA will generate is 37. The better the score the better the alignment quality
- CIGAR: a sequence of letters and numbers that represent the operations that are required to match the read to the reference. The letters are operations that are used to indicate which bases align to the reference (i.e. match, mismatch, deletion, insertion), and the numbers indicate the associated base lengths. For example 129M1I31M means that the first 129 bases match, then we have 1 insertion followed by 31 matches
- MRNM: the mate reference name
- MPOS: the mate position (1-based, leftmost)
- ISIZE: the inferred insert size
- SEQ: the raw sequence
- QUAL: the associated quality values for each position in the read
Samtools View
While the SAM alignment file from Bowtie 2 is human readable, we need a BAM alignment file for downstream analysis. A BAM file is a binary equivalent version of the SAM file, i.e. the same file in a compressed format.
We can use the samtools view command to convert our SAM file into its binary compressed version (BAM) and save it to file. Once we generate the BAM file, we don’t need to retain the SAM file anymore, we can delete it to save space.
samtools view -h -S -b \
-o SRR6344904_mapped.bam \
SRR6344904_mapped.samWe can adjust this command to ask very specific questions about our data as well. For example, we might ask how many insertions and deletions we have in our mapped reads. For this, we can use samtools view followed by some extra commands that we add with a pipe to:
- Extract only the mapped reads by removing the unmapped reads (
-F 4) - Extract the 6th field with the CIGAR string that contains information about insertions, deletions, etc (
cut -f 6) - Extract only alignments that have insertion or deletions (
grep -P '[ID]') - Count how many alignments have insertion or deletions (
wc -l)
#count total alignments
SRR6344904_mapped_sorted.bam | wc -l
#count mapped alignments with ID events
samtools view -F 4 SRR6344904_mapped_sorted.bam | \
cut -f 6 | \
grep -P '[ID]' | \
wc -l}Used options:
-h: include header in output-S: input is in SAM format-b: output BAM format-o: /path/to/output/file-F 4: exclude reads with the flag 4 (i.e. unmapped reads)-f 4: only keep reads with the flag 4 (i.e. unmapped reads)
Samtools Sort
Sorting BAM files is recommended for most down-stream analyses and is done as follows:
samtools sort \
SRR6344904_mapped.bam \
-o SRR6344904_mapped_sorted.bamSamtools Index
The samtools index command creates a new index file that allows fast look-up of the data in a sorted SAM or BAM file.
samtools index SRR6344904_mapped_sorted.bamSamtools Idxstats
The samtools idxstats command prints stats for the BAM index file but it requires an index to run.
The output is TAB delimited with each line consisting of reference sequence name, sequence length, number of mapped reads and number of unmapped reads.
samtools idxstats SRR6344904_mapped_sorted.bamSamtools depth
Samtools depth computes the read depth at each position or region
#calculate the depth per position
#header: the name of the contig or chromosome, the position, the number of reads aligned at that position
samtools depth SRR6344904_mapped_sorted.bam | head
#calculate the average coverage for all covered regions
samtools depth SRR6344904_mapped_sorted.bam | awk '{sum+=$3} END { print "Average = ",sum/NR}'
#calculate the average coverage for all regions
samtools depth -a SRR6344904_mapped_sorted.bam | awk '{sum+=$3} END { print "Average = ",sum/NR}'Notice:
- If you want to calculate the average X coverage for your genome, then you would need to divide by the total size of your genome, instead of dividing by NR in the command above.
References
Danecek, Petr, James K Bonfield, Jennifer Liddle, John Marshall, Valeriu Ohan, Martin O Pollard, Andrew Whitwham, et al. 2021. “Twelve Years of SAMtools and BCFtools.” GigaScience 10 (2). https://doi.org/10.1093/gigascience/giab008.