conda config --add envs_dirs /zfs/omics/projects/bioinformatics/software/miniconda3/envs/Bakta
Introduction
Bakta is a tool for the rapid & standardized annotation of bacterial genomes and plasmids from both isolates and MAGs (Schwengers et al. 2021). It provides dbxref-rich, sORF-including and taxon-independent annotations in machine-readable JSON & bioinformatics standard file formats for automated downstream analysis. The Bakta workflow looks as follows:
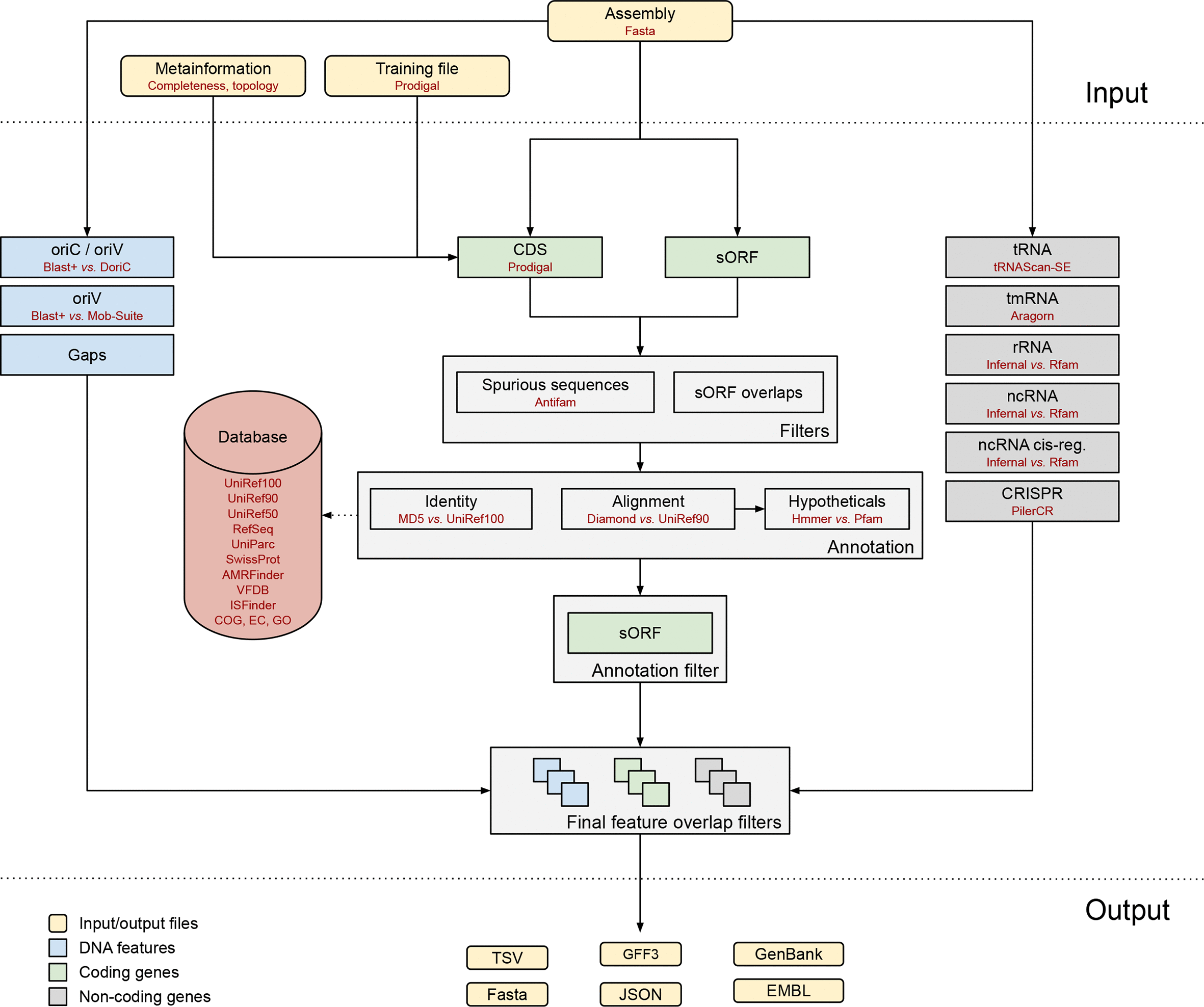
Please, note that Bacta was designed and developed to annotate isolated bacterial genomes. In particular, the database comprises bacterial protein coding genes, only. Also, there are currently no options/parameters to surpass archaeal taxonomic information to 3rd party tools of the workflow, e.g. tRNAScan-SE. Therefore, the identification of CDS and protein sequences can be still used but for functional annotation it is recommended to use archaeal-specific or domain-unspecific databases.
There also is a web-server on which you can upload your genomes or visualize bakta json files that you have already generated.
For more information, please visit the manual.
Installation
Installed on crunchomics: Yes,
- Bakta v1.9.4 is installed as part of the bioinformatics share. If you have access to crunchomics and have not yet access to the bioinformatics you can send an email with your Uva netID to Nina Dombrowski, n.dombrowski@uva.nl.
- Afterwards, you can add the bioinformatics share as follows (if you have already done this in the past, you don’t need to run this command):
If you want to install it yourself, you can run the command below — it doesn’t pin a specific version, so it will install whatever the latest release is at the time you run it.
# Install bakta
mamba create -p /zfs/omics/projects/bioinformatics/software/miniconda3/envs/bakta_1.9.4 -c conda-forge -c bioconda bakta
# Setup database
mamba activate bakta_1.9.4
bakta_db download --output /zfs/omics/projects/bioinformatics/databases/bakta --type full
mamba deactivate
# To update an existing database you can use the following code:
#bakta_db update --db <existing-db-path> [--tmp-dir <tmp-directory>]Usage
mkdir -p results/bakta
conda activate bakta_1.9.4
bakta --db /zfs/omics/projects/bioinformatics/databases/bakta/db \
--verbose \
--output results/bakta \
--prefix strainX \
--locus-tag strainX \
--threads 8 \
--force \
strainX.fasta
conda deactivateFor a complete description, please visit the manual.
Generated output
Annotation results are provided in standard bioinformatics file formats:
<prefix>.tsv: annotations as simple human readable TSV<prefix>.gff3: annotations & sequences in GFF3 format<prefix>.gbff: annotations & sequences in (multi) GenBank format<prefix>.embl: annotations & sequences in (multi) EMBL format<prefix>.fna: replicon/contig DNA sequences as FASTA<prefix>.ffn: feature nucleotide sequences as FASTA<prefix>.faa: CDS/sORF amino acid sequences as FASTA<prefix>.hypotheticals.tsv: further information on hypothetical protein CDS as simple human readable tab separated values<prefix>.hypotheticals.faa: hypothetical protein CDS amino acid sequences as FASTA<prefix>.json: all (internal) annotation & sequence information as JSON<prefix>.txt: summary as TXT<prefix>.png: circular genome annotation plot as PNG<prefix>.svg: circular genome annotation plot as SVG
The <prefix> can be set via --prefix <prefix>. If no prefix is set, Bakta uses the input file prefix.
List of Bakta options
usage: bakta [--db DB] [--min-contig-length MIN_CONTIG_LENGTH] [--prefix PREFIX] [--output OUTPUT]
[--genus GENUS] [--species SPECIES] [--strain STRAIN] [--plasmid PLASMID]
[--complete] [--prodigal-tf PRODIGAL_TF] [--translation-table {11,4}] [--gram {+,-,?}] [--locus LOCUS]
[--locus-tag LOCUS_TAG] [--keep-contig-headers] [--replicons REPLICONS] [--compliant] [--replicons REPLICONS] [--regions REGIONS] [--proteins PROTEINS] [--meta]
[--skip-trna] [--skip-tmrna] [--skip-rrna] [--skip-ncrna] [--skip-ncrna-region]
[--skip-crispr] [--skip-cds] [--skip-pseudo] [--skip-sorf] [--skip-gap] [--skip-ori] [--skip-plot]
[--help] [--verbose] [--debug] [--threads THREADS] [--tmp-dir TMP_DIR] [--version]
<genome>
Rapid & standardized annotation of bacterial genomes, MAGs & plasmids
positional arguments:
<genome> Genome sequences in (zipped) fasta format
Input / Output:
--db DB, -d DB Database path (default = <bakta_path>/db). Can also be provided as BAKTA_DB environment variable.
--min-contig-length MIN_CONTIG_LENGTH, -m MIN_CONTIG_LENGTH
Minimum contig size (default = 1; 200 in compliant mode)
--prefix PREFIX, -p PREFIX
Prefix for output files
--output OUTPUT, -o OUTPUT
Output directory (default = current working directory)
--force, -f Force overwriting existing output folder (except for current working directory)
Organism:
--genus GENUS Genus name
--species SPECIES Species name
--strain STRAIN Strain name
--plasmid PLASMID Plasmid name
Annotation:
--complete All sequences are complete replicons (chromosome/plasmid[s])
--prodigal-tf PRODIGAL_TF
Path to existing Prodigal training file to use for CDS prediction
--translation-table {11,4}
Translation table: 11/4 (default = 11)
--gram {+,-,?} Gram type for signal peptide predictions: +/-/? (default = ?)
--locus LOCUS Locus prefix (default = 'contig')
--locus-tag LOCUS_TAG
Locus tag prefix (default = autogenerated)
--keep-contig-headers
Keep original contig headers
--compliant Force Genbank/ENA/DDJB compliance
--replicons REPLICONS, -r REPLICONS
Replicon information table (tsv/csv)
--regions REGIONS Path to pre-annotated regions in GFF3 or Genbank format (regions only, no functional annotations).
--proteins PROTEINS Fasta file of trusted protein sequences for CDS annotation
--meta Run in metagenome mode. This only affects CDS prediction.
Workflow:
--skip-trna Skip tRNA detection & annotation
--skip-tmrna Skip tmRNA detection & annotation
--skip-rrna Skip rRNA detection & annotation
--skip-ncrna Skip ncRNA detection & annotation
--skip-ncrna-region Skip ncRNA region detection & annotation
--skip-crispr Skip CRISPR array detection & annotation
--skip-cds Skip CDS detection & annotation
--skip-pseudo Skip pseudogene detection & annotation
--skip-sorf Skip sORF detection & annotation
--skip-gap Skip gap detection & annotation
--skip-ori Skip oriC/oriT detection & annotation
--skip-plot Skip generation of circular genome plots
General:
--help, -h Show this help message and exit
--verbose, -v Print verbose information
--debug Run Bakta in debug mode. Temp data will not be removed.
--threads THREADS, -t THREADS
Number of threads to use (default = number of available CPUs)
--tmp-dir TMP_DIR Location for temporary files (default = system dependent auto detection)
--version show program's version number and exitReferences
Schwengers, Oliver, Lukas Jelonek, Marius Alfred Dieckmann, Sebastian Beyvers, Jochen Blom, and Alexander Goesmann. 2021. “Bakta: Rapid and Standardized Annotation of Bacterial Genomes via Alignment-Free Sequence Identification.” Microbial Genomics 7 (11). https://doi.org/10.1099/mgen.0.000685.