conda config --add envs_dirs /zfs/omics/projects/bioinformatics/software/miniconda3/envs/
conda config --add envs_dirs /zfs/omics/projects/metatools/TOOLS/miniconda3/envs/Metabolic
Introduction
Metabolic is a workflow developed by the AnantharamanLab. This software enables the prediction of metabolic and biogeochemical functional trait profiles to any given genome datasets. These genome datasets can either be metagenome-assembled genomes (MAGs), single-cell amplified genomes (SAGs) or isolated strain sequenced genomes.
METABOLIC has two main implementations, which are METABOLIC-G and METABOLIC-C. METABOLIC-G.pl allows for generation of metabolic profiles and biogeochemical cycling diagrams of input genomes and does not require input of sequencing reads. METABOLIC-C.pl generates the same output as METABOLIC-G.pl, but as it allows for the input of metagenomic read data, it will generate information pertaining to community metabolism.
Check out the manual for all usage options and more details about what the software exactly does (Zhou et al. 2022).
Installation
Available on crunchomics: Yes,
- Metabolic v4.0 is installed as part of the bioinformatics share. If you have access to crunchomics and have not yet access to the bioinformatics you can send an email with your Uva netID to Nina Dombrowski, n.dombrowski@uva.nl.
- Additionally, the GTDB r214 database that is used by metabolic is installed on the metatools share. To get access to metatools, send an email with your Uva netID to Anna Heintz Buschart, a.u.s.heintzbuschart@uva.nl.
After you were added to the bioinformatics and metatools share, you can add the conda environments that are installed in these shares as follows (if you have already done this in the past, you don’t need to run these commands):
If you want to install it yourself, you can run:
#go into folder in which to install software
cd ~/personal/software/METABOLIC_4.0
#download environmental yaml (which tells conda what software to install)
mkdir envs
wget https://raw.githubusercontent.com/AnantharamanLab/METABOLIC/master/METABOLIC_v4.0_env.yml -P envs/
#install dependencies via the environmental yaml (this env will be named METABOLIC_v4.0)
mamba env create -f envs/METABOLIC_v4.0_env.yml
#activate environment (NEEDS to be active to run the setup steps below)
conda activate METABOLIC_v4.0
#download a git clone of the METABOLIC workflow
git clone https://github.com/AnantharamanLab/METABOLIC.git
#run bash setup script (needs some time, have patience)
cd METABOLIC
bash run_to_setup.sh
Note
The command bash run_to_setup.sh installs some public databases, which might be useful for other things. These are:
- The kofam database: KOfams are a customized HMM database of KEGG Orthologs (KOs). The KO (KEGG Orthology) database is a database of molecular functions represented in terms of functional orthologs and is useful to assign functions to your proteins of interest. The script will download the database from scratch and you will therefore always have the newest version installed when installing METABOLIC.
- The dbCAN2 database: A database that can be used for carbohydrate-active enzyme annotation. The script downloads dbCAN v10.
- The Meropds database: A database for peptidases (also termed proteases, proteinases and proteolytic enzymes) and the proteins that inhibit them. The script will download the most recent version from the internet.
Additionally to these databases, metabolic makes use of the gtdb database. If desired, you can install the GTDB database yourself as follows:
#go into the folder into which you want to download the database
cd /path/to/target
#Manually download the latest reference data
#Check, if you work with the latest database. When installing Metabolic, you might get some pointers how to set up gtdb and we recommend to follow those
wget https://data.gtdb.ecogenomic.org/releases/release207/207.0/auxillary_files/gtdbtk_r207_v2_data.tar.gz
#Extract the archive to a target directory:
#change `/path/to/target/db` to whereever you want to install the db
tar -xvzf gtdbtk_r207_v2_data.tar.gz -c "/path/to/target/db" --strip 1 > /dev/null
#cleanup
rm gtdbtk_r207_v2_data.tar.gz
#while the conda env for METABOLIC is activate link gtdb database
#change "/path/to/target/db" to wherever you downloaded the database
conda env config vars set GTDBTK_DATA_PATH="/path/to/target/db"
#reactivate env for the variable to be recognized
conda deactivate
conda activate METABOLIC_v4.0Usage example
METABOLIC has two scripts:
- METABOLIC-G.pl: Allows for classification of the metabolic capabilities of input genomes.
- METABOLIC-C.pl: Allows for classification of the metabolic capabilities of input genomes, calculation of genome coverage, creation of biogeochemical cycling diagrams, and visualization of community metabolic interactions and contribution to biogeochemical processes by each microbial group.
Input:
- Nucleotide fasta files (use
-in-gnin the perl script) - Protein faa files (use
-inin the perl script) - Illumina reads (use
-rflag in the metabolic-c perl script) provided as un-compressed fastq files. This option requires you to provide the full path to your paired reads. Note that the two different sets of paired reads are separated by a line return (new line), and two reads in each line are separated by a “,” but not ” ,” or ” , ” (no spaces before or after comma). Blank lines are not allowed - Nanopore/PacBio long-reds (use
rtogher with-st illumina/pacbio/pacbio_hifi/nanoporeto provide information about the sequencing type) - If you provide the raw reads, the should be given as a text file with the absolute paths that looks something like this:
#Read pairs:
/path/to/your/reads/file/SRR3577362_sub_1.fastq,/path/to/your/reads/file/SRR3577362_sub_2.fastq
/path/to/your/reads/file/SRR3577362_sub2_1.fastq,/path/to/your/reads/file/SRR3577362_sub2_2.fastqImportant:
- Ensure that the fasta headers of each “.fasta” or “.faa” file is unique (all fasta or faa files will be concatenated together to make a “total.fasta” or “total.faa” file; be sure that all sequence headers are unique)
- that your file names do not contain spaces (suggest to only use alphanumeric characters and underscores in the file names)
- be sure that in the genomes folder, only the genomes are placed but not other files, for example, non-genome metagenomic assemblies, since METABOLIC will take in all the files within the folder as genomes.
- If you want to use METABOLIC-C, only “fasta” files and the “-in-gn” flag are allowed to perform the analysis correctly.
Some example files for testing can be found in METABOLIC_test_files/. The steps below show how to get the files and also includes some sanity checks that can be useful to test, whether your files are suitable to be used by metabolic:
Preparing the input files
We start with downloading proteins from two genomes and do some cleaning, here we:
- Make sure the file header is concise and does not have ANY spaces and that ideally uses a ‘-’ (or any other unique delimiter) to separate the genome ID from the protein ID. Also avoid any unusual symbols, such as |, (, ), {, }…
- Add not only the protein ID but also the genome ID sequence header
- If you have a concise header + the bin ID in the header, it is easy for METABOLIC to concatenate the protein sequences of your genomes into one single file and still easily know from what genome the sequence originally came from
mkdir -p results/faa/renamed
#download some example genomes from NCBI
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/008/085/GCA_000008085.1_ASM808v1/GCA_000008085.1_ASM808v1_protein.faa.gz -P results/faa/
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/017/945/GCA_000017945.1_ASM1794v1/GCA_000017945.1_ASM1794v1_protein.faa.gz -P results/faa/
gzip -d results/faa/*gz
#view the header of one file
#we see the header looks like this: >ABU81185.1 translation initiation factor aIF-2 [Ignicoccus hospitalis KIN4/I]
#this is too long and the extra characters, i.e. spaces, can disrupt downstream analysis, so let's fix that first
head results/faa/GCA_000017945.1_ASM1794v1_protein.faa
#remove everything after the space in the fasta headers
for i in results/faa/*faa; do
filename=$(basename $i .faa)
sed '/^>/ s/ .*$//g' $i > results/faa/${filename}_temp.faa
done
#afterwards, the header looks like this; >ABU81185.1
head results/faa/GCA_000017945.1_ASM1794v1_protein_temp.faa
#next, we add the filename into the fasta header and store them in our new folder
#this allows us to combine all genomes into 1 file has the benefit that you only need to run hmmsearch 1x
#for this to work proberly its a good practice to add the genomeID into the fasta header so that you can easily distinguish from where each protein originally came
for i in results/faa/*_temp.faa; do
filename=$(basename $i _temp.faa | cut -f1 -d ".")
awk -v fname="$filename" '/>/{sub(">","&"fname"-")}1' $i > results/faa/renamed/$filename.faa
done
#afterwards, the header looks like this; >ABU81185.1
#notice: how using basename with cut made our filename a bit shorter?
head results/faa/renamed/GCA_000017945.faa
#cleanup the temporary files we made
rm results/faa/*temp.faa
TipTip: How does the sed command work
Let’s look into how this code works:
for i in results/faa/*faa; do
filename=$(basename $i .faa)
sed '/^>/ s/ .*$//g' $i > results/faa/${filename}_temp.faa
donefor i in results/faa/*faa; do: This line starts a loop that iterates over each file in the directoryresults/faa/that ends with the extension.faa. The loop variableiwill hold the path of each file in turn during each iteration.filename=$(basename $i .faa): Inside the loop, this line extracts the base name of the current file ($i) by removing the directory path and the.faaextension. It assigns this base name to the variablefilename.sed '/^>/ s/ .*$//g' $i: This line uses thesedcommand to edit the contents of the current file ($i). Let’s break down thesedcommand:'/^>/ s/ .*$//g': This is a regular expression pattern thatsedwill use to find and replace text in the file.^>: This part of the pattern matches lines that start with>.s/ .*$//g: This part of the pattern replaces any space character and everything after it on lines that match the pattern with nothing (i.e., it removes everything after the first space). Here, thesstands for substitution, and the.*$matches any character (.) until the end of the line ($).
$i: This is the file thatsedwill operate on, which is the current file in the loop.
- We then store the output to a new file with
> results/faa/${filename}_temp.faa. This makes use of the filename variable to flexibly edit the filename.
done: This marks the end of the loop. So, in summary, this code loops through each file in the directoryresults/faa/with the.faaextension, extracts the base name of each file, and then edits each file in-place to remove any text after the first space on lines that start with>.
TipTip: How does the awk command work
Let’s look into how this code works:
for i in results/faa/*_temp.faa; do
filename=$(basename $i _temp.faa | cut -f1 -d ".")
awk -v fname="$filename" '/>/{sub(">","&"fname"-")}1' $i > results/faa/renamed/$filename.faa
donefor i in results/faa/*_temp.faa; do: This line initiates a loop that iterates over each file in the directoryresults/faa/with the_temp.faaextension. During each iteration, the variableiholds the path of the current file.filename=$(basename $i _temp.faa | cut -f1 -d "."): Inside the loop, this line extracts the base name of the current file ($i) by removing the directory path and the_temp.faaextension usingbasename, and then it usescutto split the filename at the “.” character and extracts the first part. The extracted base name is stored in the variablefilename.awk -v fname="$filename" '/>/{sub(">","&"fname"-")}1' $i > results/faa/renamed/$filename.faa: This line utilizes theawkcommand to process the contents of the current file ($i). Let’s break down theawkcommand:-v fname="$filename": This option passes the value of the shell variablefilenametoawkas anawkvariable namedfname./>/: This part of the pattern matches lines that contain>.{sub(">","&"fname"-")}: This action performs a substitution on lines that match the pattern:sub(">","&"fname"-"): This substitutes the>character with itself (&) followed by the value offnameand a hyphen (-). So, it’s essentially appending the value offnamefollowed by a hyphen after the>character.
1: This is a condition that always evaluates to true, triggering the default action ofawk, which is to print the current line.$i: This is the file thatawkwill operate on.> results/faa/renamed/$filename.faa: This redirects the output ofawkto a new file with the same base name as the original file ($filename.faa), but located in theresults/faa/renamed/directory.
So, in summary, this code loops through each file in the directory results/faa/ with the .faa extension, extracts the base name of each file, and then uses awk to modify each file’s contents. It appends the base name followed by a hyphen after any line that starts with >, and saves the modified content to a new file in the results/faa/renamed/ directory with the same base name as the original file.
Run the G mode with faa files as input:
Important:
- Metabolic combines the faa files and generates a file named
total.faain the folder with the fasta sequences, if you re-run Metabolic, ensure that you first delete that file! - As described above, metabolic runs in different modes and also can take fasta files or raw reads as input. For details on how to do that, please visit the manual
mkdir results/metabolic/
conda activate METABOLIC_v4.0
perl /zfs/omics/projects/bioinformatics/software/metabolic/v4.0.0/METABOLIC-G.pl \
-in results/faa/renamed/ \
-t 20 \
-o results/metabolic
#get a table with the raw results
#this table is good to use for sanity checks and for example test if your protein is really absent
cat results/metabolic/intermediate_files/Hmmsearch_Outputs/*hmmsearch_result.txt | sed -n '/^#/!p' > results/metabolic/hmmer_raw.txt
conda deactivateGenerated outputs (for more detail, check the manual):
- All_gene_collections_mapped.depth.txt: The gene depth of all input genes (METABOLIC-C only)
- Each_HMM_Amino_Acid_Sequence/: The faa collection for each hmm file
- intermediate_files/: The hmmsearch, peptides (MEROPS), CAZymes (dbCAN2), and GTDB-Tk (only for METABOLIC-C) running intermediate files
- KEGG_identifier_result/: The hit and result of each genome by Kofam database
- METABOLIC_Figures/: All figures output from the running of METABOLIC
- METABOLIC_Figures_Input/: All input files for R-generated diagrams
- METABOLIC_result_each_spreadsheet/: TSV files representing each sheet of the created METABOLIC_result.xlsx file
- MW-score_result/: The resulted table for MW-score (METABOLIC-C only)
- METABOLIC_result.xlsx: The resulting excel file of METABOLIC
Required/Optional flags: (for a detail explanation, please read the manual)
-in-gn[required if you are starting from nucleotide fasta files] Defines the location of the FOLDER containing the genome nucleotide fasta files ending with “.fasta” to be run by this program-in[required if you are starting from faa files] Defines the location of the FOLDER containing the genome amino acid files ending with “.faa” to be run by this program-r[required] Defines the path to a text file containing the location of paried reads-rt[optional] Defines the option to use “metaG” or “metaT” to indicate whether you use the metagenomic reads or metatranscriptomic reads (default: ‘metaG’). Only required when using METABOLIC-C-st[optional] To use “illumina” (for Illumina short reads), or “pacbio” (for PacBio CLR reads), or “pacbio_hifi” (for PacBio HiFi/CCS genomic reads (v2.19 or later)), or “pacbio_asm20” (for PacBio HiFi/CCS genomic reads (v2.18 or earlier)), or “nanopore” (for Oxford Nanopore reads) to indicate the sequencing type of metagenomes or metatranscriptomes (default: ‘illumina’; Note that all “illumina”, “pacbio”, “pacbio_hifi”, “pacbio_asm20”, and “nanopore” should be provided as lowercase letters and the underscore “_” should not be typed as “-” or any other marks)-t[optional] Defines the number of threads to run the program with (Default: 20)-m-cutoff[optional] Defines the fraction of KEGG module steps present to designate a KEGG module as present (Default: 0.75)- -kofam-db [optional] Defines the use of the full (“full”) or reduced (“small”) KOfam database by the program (Default: ‘full’). “small” KOfam database only contains KOs present in KEGG module, using this setting will significantly reduce hmmsearch running time.
-tax[optional] To calculate MW-score contribution of microbial groups at the resolution of which taxonomical level (default: “phylum”; other options: “class”, “order”, “family”, “genus”, “species”, and “bin” (MAG itself)). Only required when using METABOLIC-C-p[optional] Defines the prodigal method used to annotate ORFs (“meta” or “single”)(Default: “meta”)-o[optional] Defines the output directory to be created by the program (Default: current directory)
Addon: Verifying your results
In bioinformatics, it’s important to look at your results with a critical eye. This is particularly crucial in functional assignments, where reliance on automated tools may yield inaccuracies. Let’s delve into this with a practical example:
Consider Ignicoccus hospitalis (GCA_000017945), a genome purported to lack an ATPase according to our analysis. However, upon closer inspection of the literature, we discover that Ignicoccus does indeed possess an ATPase. This dissonance underscores the importance of critically evaluating computational findings.
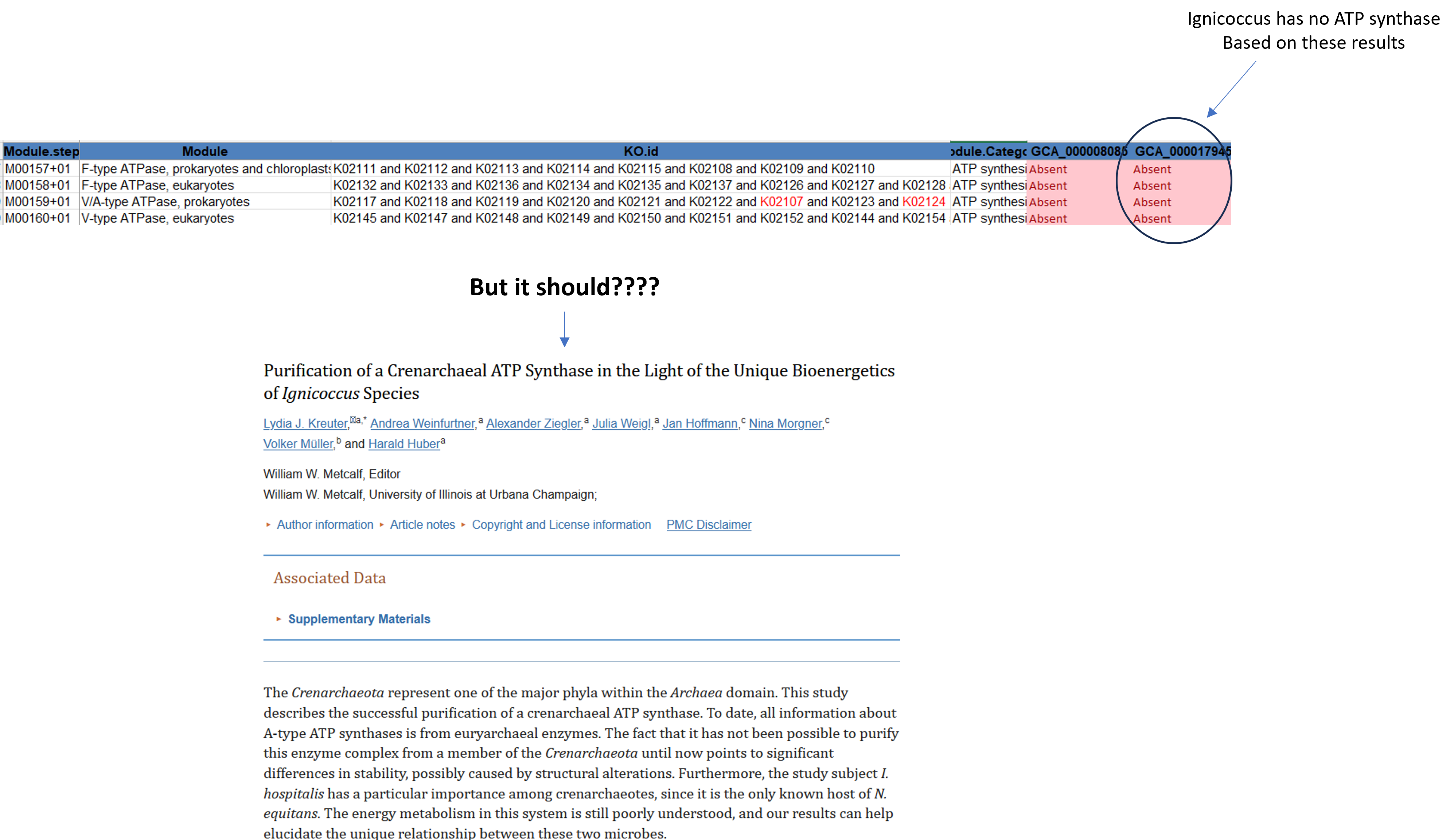
If we make a list of the IDs that are required to make up an V/ATPase in prokaryotes and search the raw hmm results, we actually see that most KOs are found albeit some with lower bitscores.
grep -f atp_list results/metabolic/hmmer_raw.txt | grep "GCA_000017945"For each hit, Metabolic uses specific bitscore cutoff values to decide whether it sees a hit as present or absent. This approach usually works well but can be too stringent as seen in our example. Due to that we can not be completely sure if a gene/module/pathway that is labelled as absent is truly absent.
So what do we do about this when we analyse our own genomes? There are a few things that can help:
- Utilize multiple annotation tools and compare their outcomes. Discrepancies among results can flag potential errors or ambiguities.
- Review raw output data (if possible) to assess the reliability of individual predictions. In the section about Hmmsearch you also find some code how you could run a hmmsearch against the KOs yourself.
- Instead of relying solely on one database, cross-reference findings from multiple sources such as Pfam, PGAP, COG, or arCOG. Consistency across databases enhances confidence in the results.
- Compare your findings against functional annotations of closely related genomes. For example, you can check the literature or check whether your organisms is part of the KEGG genome database.
- Validate specific protein functions using specialized tools like Blast or interproscan
References
References
Zhou, Zhichao, Patricia Q. Tran, Adam M. Breister, Yang Liu, Kristopher Kieft, Elise S. Cowley, Ulas Karaoz, and Karthik Anantharaman. 2022. “METABOLIC: High-Throughput Profiling of Microbial Genomes for Functional Traits, Metabolism, Biogeochemistry, and Community-Scale Functional Networks.” Microbiome 10 (1): 33. https://doi.org/10.1186/s40168-021-01213-8.